바이오의약품 수출지원, 글로벌 백신 허브 구축 등을 위해 허가품목이 없는 국내 CMO 업체도 GMP 평가를 받을 수 있게 됐다.
식품의약품안전처는 허가(신청) 품목이 없이 고객사의 수주를 받아 바이오의약품을 전문적으로 생산하는 CMO에 대해 ‘바이오의약품 전문수탁 제조업체 GMP 평가 절차“ 공무원 지침서를 개정했다고 21일 발표했다.
지침서에는 GMP 업무 수행에 관한 담당자의 역할과 책임, 세부처리절차 등을 명시했다.
식약처는 지침서 개정 배경으로 “바이오의약품 시장 확대와 함께 수탁 제조업체(CMO)가 크게 성장함에 따라 바이오의약품의 제조를 위탁하고자 하는 업체(해외 제약업체 포함)는 위ㆍ수탁 계약에 앞서 CMO의 의약품 제조 및 품질관리기준(GMP)을 객관적으로 확인하기 위하여 CMO에 대한 정부 기관의 GMP 평가결과를 요구가 있다”면서 “허가(신청) 품목이 없는 CMO의 경우 식약처장(지방청장 포함)의 GMP 평가를 받지 않으므로 이를 제시하지 못하여 제조 수탁에 어려움이 있다”고 설명했다.
이에 따라 식약처는 허가(신청) 품목이 없이 고객사의 수주를 받아 바이오의약품을 전문적으로 생산하는 CMO에 대한 GMP 업무 수행에 관한 담당자의 역할과 책임, 세부처리절차 등을 문서화 했다. 식약처는 “업무의 정확성, 일관성 및 투명성을 확보하기 위한 것”이라고 밝혔다.
식약처는 바이오의약품 CMO에 대한 GMP 실시상황 평가는 최소 3개 제조단위 이상의 수탁제조공정(원료 및 완제의약품, 일부 공정 포함) 실적을 제출받아 실시한다. 단 임상시험용 생산의 경우는 최소 1개 제조단위 이상의 수탁제조공정 실적과 향후 2개 제조단위 이상의 추가 생산계획서(예: 공정밸리데이션, 세척밸리데이션 계획서 등)를 제출받아 실시한다.
구체적으로 살펴보면 바이오의약품 품목허가(신청) 미보유 업체 또는 바이오의약품 품목허가(신청)보유 업체 중 특정 바이오의약품 제제에 대한 전문수탁을 희망하는 업체로 한정했다. 다만 ‘의약품 GMP 적합판정서’를 받은 바이오의약품 동일 제제는 제외했다. 바이오의약품 동일 제제 기준은 6개 군으로 나눠 제제를 분류했다.<표 참조>
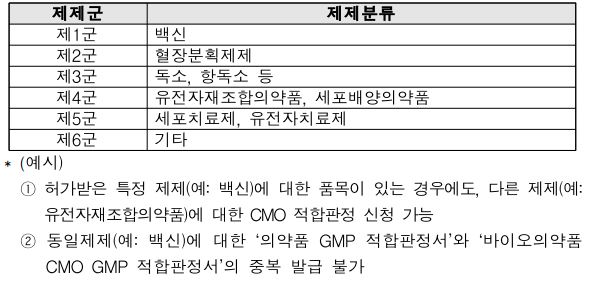
GMP 평가는 CMO가 생산하고자 하는 바이오의약품 제제별, 제형별, 제조 방법별로 평가하되 제출자료에 대한 평가 및 현장 실태 조사를 실시한다.
전문수탁 제조업체에서 제조한 제품이 향후 바이오의약품의 품목허가를 신청하면 수행된 GMP 평가는 적용되지 않으며 신규품목허가 신청에 대한 GMP 실시상황 평가를 실시해야 한다.